|
单细胞测序技术服务 靶向单细胞测序(lncRNA&mRNA) 单细胞测序 |
|
蛋白表达定量 DIA定量蛋白质组学 Label free非标定量 |
蛋白修饰定量 N-糖基化蛋白组学 O-GlcNAc修饰蛋白质组学 |
|
Ribo-seq Ribo seq(ribosome profiling) |
核糖体-新生肽链复合物(RNC) RNC联合 circRNA芯片 RNC联合 lncRNA芯片 RNC-seq |
|
NGS测序技术服务 环状DNA测序(eccDNA测序) |
PCR技术服务 环状DNA PCR技术服务 |
表观遗传学研究的是基因序列不发生改变的可遗传改变,这种可遗传改变调控基因表达变化,是基因表达转录调控的另一个层面。因此,DNA序列作用仅在于承载遗传信息,而表观遗传的机制对真核生物的发育却能起到调控的作用(图1)。
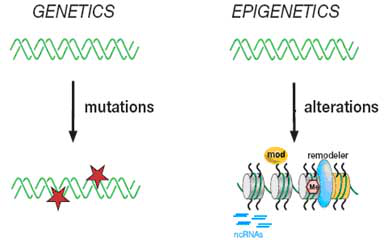
图1 遗传和表观遗传
遗传:DNA模板(绿色螺旋标注)的突变(红星标注)。表观遗传:通过(1)组蛋白修饰(mod)、(2)染色质重塑(remodeler)、(3)组蛋白变异(yellow nucleosome)、(4)DNA甲基化(Me)和非编码RNA等方式在染色质结构上发生的变异。这些表观遗传的标志可通过细胞分裂而遗传到下一代的,并且不断积累最终决定细胞的表型。
1994年,Holliday将表观遗传定义为不基于DNA序列的改变的细胞核遗传(Holliday 1994)。2006年,表观遗传被定义为染色质模板发生改变的总和,这些改变使得相同序列的基因组却呈现出不同的基因表达和沉默模式,并且这种改变能代代相传(Allis et.at. 2006)。
核小体是染色质的基本结构单元(Kornberg 1974)。而核小体是由一段DNA链缠绕组蛋白八聚体而形成(图2)。染色质/DNA-核小体多聚物是染色体的构件(Luger et at. 1997)。染色质的结构不是固定不变的,由于染色质包装折叠的松紧程度不同使染色质呈现两种形态(图3),高度螺旋、折叠紧密的染色质称为异染色质;染色质伸展、折叠疏松的称为常染色质,基因表达发生在常染色质内(Allis et at. 2006)。
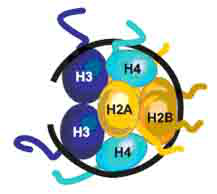
图2 核小体的结构
左图:2.8Å分辨率下的核小体模型;右图:八聚体缠绕一段DNA(黑线)的模式图。首先是H3/H4四聚体结合到DNA上,然后与两个H2A/H2B二聚体结合,便形成核小体。八个组蛋白形成圆形结构域,即核小体核心,每个组蛋白的N末端伸出核小体核心,即组蛋白的尾部。
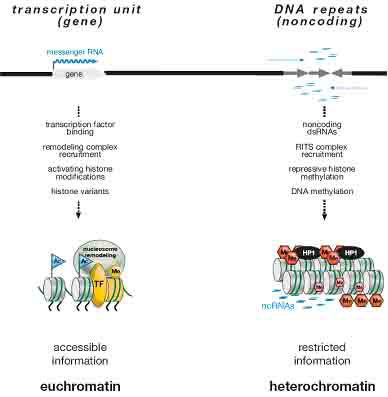
图3 常染色体和异染色体结构的区别
图中概况了常染色体和异染色体的常见的区别,包括:产生的转录本不同、招募的DNA结合蛋白不同(例如:转录因子)、染色质相关蛋白及复合物不同、组蛋白共价修饰不同以及组蛋白变异体不同等。
表观遗传在正常的发育和细胞生长过程中起作用,并且在基因、环境与疾病的关系之间也可能起到至关重要的作用。表观遗传异常已经被发现是癌症、遗传病、儿科疾病以及自身免疫性疾病和衰老等的成因。
表观遗传的机制包括(但不限于):DNA甲基化(胞嘧啶5’位碳的甲基化)、组蛋白的翻译后修饰、组蛋白的变异、染色质重塑(结构发生改变)、基因印记以及RNA干扰(非编码RNA或者基因沉默)等等(Jenuwein 2006)。
DNA甲基化(CpG岛)及其作用
DNA甲基化是一种表观遗传事件,其通过改变基因的表达来影响细胞的功能。甲基化是指在CpG双核苷酸位点的胞嘧啶上加上一个甲基基团,这一反应是由DNA甲基转移酶(DNMT)的催化完成的。
5’-甲基胞嘧啶是在DNA甲基转移酶(DNMT1,3a,3b)的催化作用下,甲基基团(CH3)从S-腺苷甲硫氨酸(SAM)转移到胞嘧啶的5位碳原子上。DNA甲基化无一例外的发生在CpG位点,CpG岛富含CpG位点,其甲基化状态对于维持正常的胚胎发育以及基因组印记、X-染色体失活都是必需的,具有重要的生物学意义。
在基因组大多数序列中,CpG双核苷酸出现频率并不多;但是在CpG岛中却富含CpG,每个CpG岛长度约1kb,其中CpG双核苷酸的出现频率如预期一样高。整个基因组中约有45,000个CpG岛,大多数CpG岛位于基因的启动子及第一外显子区域。
在正常细胞中,CpG岛处于非甲基化状态。DNA甲基化的改变与疾病密切相关。
CpG岛多位于基因的5’端,是调控下游基因表达的分子开关。位于5’端的CpG岛通常处于非甲基化状态,便于这些基因表达。
在一些癌症中,肿瘤抑制基因5’端的CpG岛甲基化,引起基因表达关闭(例如,肿瘤抑制基因(p53或p16)附近的CpG岛的甲基化通常与这些基因的沉默相关)。DNA甲基化使基因沉默的机制是肿瘤抑制基因失活的主要机制之一。
CpG岛与75%的人类基因的表达调控有关(loshikhes and Zhang 2000)。CpG岛通常是GC含量>0.55(预期值为>0.5)、长度为大于300bp(不过很少有小于500bp的)的一段基因组序列(Aerts et al.2004)。CpG岛区域总是保持非甲基化状态(Rollins et al.2006)。
DNA甲基化是DNA复制后在胞嘧啶上发生甲基化的过程。在高等生物中,从植物到人类,甲基化能够保护DNA不被内切酶降解,并且在基因表达的调控过程中具有至关重要的作用,这些功能使得甲基化在正常的发育和生物学功能中起着重要的作用。
CpG位点的胞嘧啶要么甲基化要么非甲基化。当DNA一条链的CpGs被甲基化时,识别半甲基化CpG位点的DNA甲基转移酶则使互补链的CpGs甲基化,最终DNA双链都被甲基化。在DNA复制过程中,甲基化或非甲基化的状态会遗传到新产生的两条DNA分子上。DNA甲基化是基因组DNA的一种标志,并且这种标志可代代相传。
有人认为胞嘧啶的甲基化能够使基因组易变区的基因表达维持在一定的水平上,DNA重复序列及转座子的积累会导致基因组增大,而甲基化能够缓冲基因组增大产生的影响(Rollins et.al. 2006)。这一观点与已有的研究具有一致性,已有的研究表明在具有大的基因组(>5×108bp)的生物中,存在甲基化的DNA(Kidwell 2002)并且表达DNA甲基转移酶1、3的基因家族(Goll and Bestor 2005),而小基因组的真核生物中缺乏DNA甲基转移酶的基因。
DNA甲基化是一种重要的调控转录的表观遗传机制。在维持正常细胞的生理活动中DNA甲基化起着重要的作用,甲基化模式发生改变有可能导致癌症的发生发展。癌症的发生发展过程是由基因功能异常积累引发的。在1993年,首次报导了DNA甲基化异常能够引起肿瘤抑制基因的沉默。最近几年,DNA甲基化在癌症发生过程中的作用已经成为值得关注的研究热点。各种癌症以及发育过程中DNA甲基化状态通常会发生改变。在所有的表观遗传修饰中,研究最为广泛的是肿瘤抑制基因(例如BRCA1、hMLH1、p16INK4a和VHL)启动子区域高甲基化,这种高甲基化抑制转录从而使基因沉默。不过,基因组整体低水平的甲基化也被认为是癌症发生的原因。甲基化机制及其调控机制的新信息使许多调控蛋白及酶类得以发现(Das and Singal 2004)。
肿瘤细胞中DNA甲基化异常(基因组整体甲基化水平低而区域特异性甲基化水平高)出现的频率很高。基因组整体甲基化水平降低会导致染色体的不稳定性,而区域特异性甲基化水升高与肿瘤抑制基因的失活密切相关。临床前及临床研究表明:食物中生物活性的成分的防癌功效,部分原因可能与其改变了DNA甲基化模式有关(Davis and Uthus 2004)。
目前已知,衰老和慢性炎症都会引起DNA甲基化。有许多致癌物不会引起基因变异,但却能引起DNA甲基化发生异常。DNA甲基化是DNA分子的一种标记,在传代过程中不会丢失。然而,几年前才开始对DNA的甲基化标记在传代过程中的保真性进行深入分析。研究人员已对CpG位点的甲基化或非甲基化状态在遗传过程中的保真性进行了计算,而更为重要的是要分析这种保真性在肿瘤细胞中发生改变的可能性(Ushijima et al. 2003)。
组蛋白修饰及其对基因表达的作用
组蛋白修饰是位点特异的、可逆的修饰,例如乙酰化、甲基化、磷酸化、泛素化及苏素化(Peterson and Laniel 2004, Allis et al. 2006)。在表观遗传机制中,组蛋白尾部的修饰是多样化的,到目前为止,已经描绘出50多种标志性的组蛋白尾部修饰(Jenuwein 2006)(图4)。

图4 组蛋白尾部修饰位点
组蛋白N末端尾部包括大部分已知的共价修饰位点。也有一部分修饰发生在球形结构域(方框标注)。一般来说,激活标志包括乙酰化(蓝色的AC小旗)、精氨酸甲基化(还色Me六边形)和一些赖氨酸甲基化,例如H3K4和H3K36(绿色Me六边形)。在球形结构域上的H3K79起到抵抗基因沉默的作用。抑制标志有H3K9、H3K27和H4K20(红色Me六边形)。图中,绿色=激活标志,红色=抑制标志。
在染色质形成过程中,组蛋白修饰是可逆的,组蛋白修饰影响基因表达。当染色质处于松散状态,基因表达开关打开;当染色质处于致密状态,基因表达开关关闭(Rodenhiser and Mann 2006)。为了调控一些与DNA相关的生物进程,染色质会发生重塑,并有以下三种方式:一种是通过ATP依赖的核小体重塑机制动员或移开核小体组蛋白(Smith and Peterson 2005);第二种是通过组蛋白的转录后调控改变染色质的结构(Strahl and Allis 2000);第三种是通过特定的组蛋白的置换(Henikoff and Ahmad 2005, Cavalli 2006)。
组蛋白(与启动子区域结合的H3和H4)的乙酰化能松弛组蛋白-DNA结合,有利于基因转录,这一过程与乙酰化组蛋白招募含有乙酰化赖氨酸结合模块(溴区结构域, Bromodomain)的转录激活因子密切相关。在许多真核生物的转录因子中都能找到这种溴区结构域(Winston and Allis 1999)。
表观遗传修饰的研究涉及组蛋白氨基端尾部的赖氨酸的乙酰化或去乙酰化,而乙酰化或去乙酰化受组蛋白乙酰转移酶(HATs)和组蛋白去乙酰化酶(HDACs)调控。组蛋白乙酰化和去乙酰化维持平衡状态对于正常细胞生长至关重要(Waterborg 2002)。在许多肿瘤中,HATs和HDACs在结构或者表达水平上都会发生改变(Timmermann et al. 2001, Jones and Baylin 2002,……)。
组蛋白甲基化要么导致转录抑制,要么导致转录激活(Peterson and Laniel 2004, Brinkman et al. 2006, Allis et al. 2006,……)。H3K27me3(位于基因间区和沉默基因的编码区)与转录抑制相关,而另外一些组蛋白甲基化,例如H3K4me3(位于启动子区)则与转录激活相关。研究已表明H3 K9的甲基化(H3K9me3)是基因沉默的标志,但存在于基因编码区的H3K9me3似乎与基因激活有关(Brinkman et al. 2006)。
基因间区、编码区和启动子区位于染色质的不同区段。然而,一些新近的研究结果却对异染色质和常染色质泾渭分明的观点提出了质疑,并提示了:常染色质与异染色质之间通过相互依赖的模式,或者通过各自独立的修饰模式,最终共同发出基因表达与否的信号(Brinkman et al. 2006)。H3K27me3明显存在于基因间区和沉默基因的编码区,但H3K27me3也与一些激活基因的编码区有关联;与此相似的是,组蛋白H3/H4的乙酰化和H3K4me3富集于启动子区,但它们却并非基因转录的标志(Brinkman et al. 2006)。另外,H3K9me3不仅仅存在于失活的X染色体中,也位于激活基因的编码区(Brinkman et at. 2006)。对含有染色质结构域的蛋白的招募可以解释一些标志性的甲基化事件(Flanagan et al. 2005, Becker 2006)。
组蛋白甲基化和组蛋白乙酰化共同作用参与转录的沉默(图5),这便提供了一个分子网络用以解释在细胞分裂和分化期间,异染色质是如何形成和维持(Czermin and Imhof 2003)。目前已经有六个组蛋白甲基化位点得到了确认:五个在组蛋白H3上(K4、K9、K27、K36、和K79);一个在组蛋白H4上(K20)(表1)。一般认为,H3K4、H3K36和H3K79与转录激活相关,而其他的则与转录抑制相关。下表列出了已报导的组蛋白的修饰以及这些修饰是激活还是抑制基因的转录。值得注意的是,泛素化可以激活(H2B)也可以抑制(H2A)基因的转录;而苏素化则是抑制基因的转录(Allis et al. 2006)。
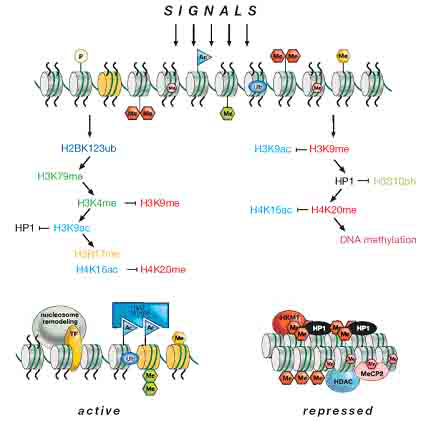
图5 染色质修饰间的协调作用
转录激活的常染色体(左边)同转录抑制的异染色体(右边)之间的相互转换,期间涉及到一系列协调作用的染色质修饰。例如,转录激活的过程中伴随着核小体的重塑和组蛋白的变异体(黄色,即H3.3)替换核心组蛋白等。

表1. 组蛋白上的转录标志
组蛋白修饰、DNA甲基化和癌症的关系
组蛋白修饰和DNA甲基化是细胞核功能的一部分。基因组的稳定性和基因表达的正确性有赖于DNA甲基化和组蛋白修饰模式的建立。而DNA甲基化和组蛋白修饰的正常模式被打破是癌症的一个特征。
已经被广泛接受的观点是:MDB蛋白通过招募组蛋白去乙酰化酶及改变染色质结构的染色质重塑活动使得DNA甲基化,引起基因沉默。
尽管组蛋白的修饰(在H3的第四位赖氨酸的乙酰化和甲基化)通常会激活基因表达,但事实上基因表达的调控有多个水平,包括H3的第17位精氨酸的甲基化、组蛋白H3.3的变异体的作用以及核小体重塑等(Esteller and Almouzni 2005)。肿瘤细胞中表观遗传修饰的发生异常的模式如下:重复序列呈现低甲基化状态并伴随着H4K16的单乙酰化和H4K20的三甲基化的丢失,与此同时,肿瘤抑制基因启动子区域的CpG岛高甲基化,并伴随着组蛋白乙酰化及H3K4三甲基化的丢失(Esteller and almouzni 2005)。
表观遗传的改变在癌症形成早期有着重要的作用,能取代在癌症发展早期发挥作用的遗传变异。因此,通过表观遗传调控干/祖细胞有可能成为癌症风险评估和化疗效果的一项重要目标(Feinberg et al. 2003)。
, i-font-family: Arial; mso-asc: Arial">)。另外,H3K9me3不仅仅存在于失活的X染色体中,也位于激活基因的编码区(Brinkman et at. 2006)。对含有染色质结构域的蛋白的招募可以解释一些标志性的甲基化事件(Flanagan et al. 2005, Becker 2006)。
组蛋白甲基化和组蛋白乙酰化共同作用参与转录的沉默(图5),这便提供了一个分子网络用以解释在细胞分裂和分化期间,异染色质是如何形成和维持(Czermin and Imhof 2003)。目前已经有六个组蛋白甲基化位点得到了确认:五个在组蛋白H3上(K4、K9、K27、K36、和K79);一个在组蛋白H4上(K20)(表1)。一般认为,H3K4、H3K36和H3K79与转录激活相关,而其他的则与转录抑制相关。下表列出了已报导的组蛋白的修饰以及这些修饰是激活还是抑制基因的转录。值得注意的是,泛素化可以激活(H2B)也可以抑制(H2A)基因的转录;而苏素化则是抑制基因的转录(Allis et al. 2006)。
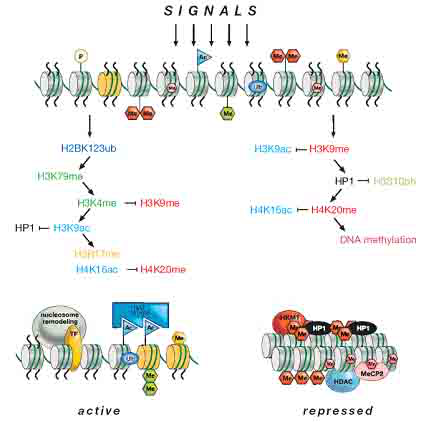
图5 染色质修饰间的协调作用
转录激活的常染色体(左边)同转录抑制的异染色体(右边)之间的相互转换,期间涉及到一系列协调作用的染色质修饰。例如,转录激活的过程中伴随着核小体的重塑和组蛋白的变异体(黄色,即H3.3)替换核心组蛋白等。


