|
单细胞测序技术服务 靶向单细胞测序(lncRNA&mRNA) 单细胞测序 |
|
蛋白表达定量 DIA定量蛋白质组学 Label free非标定量 |
蛋白修饰定量 N-糖基化蛋白组学 O-GlcNAc修饰蛋白质组学 |
|
Ribo-seq Ribo seq(ribosome profiling) |
核糖体-新生肽链复合物(RNC) RNC联合 circRNA芯片 RNC联合 lncRNA芯片 RNC-seq |
|
NGS测序技术服务 环状DNA测序(eccDNA测序) |
PCR技术服务 环状DNA PCR技术服务 |
相关服务
相关产品
相关资源
表观遗传学与疾病 表观遗传调控与肿瘤形成的分子机制DNA甲酰基胞嘧啶修饰(5fc)是DNA去甲基化过程中的中间产物,也被称为第七碱基,在生物体中稳定存在,在表观遗传中发挥着重要作用。研究报道,5fc在增强子中高度富集,这暗示5fc调控基因的转录和表达。5fc可以特异的结合特定的蛋白,参与到表观遗传调控。同时5fc可以影响DNA的双螺旋结构,有研究显示,5fc在前列腺癌的发生发展过程中起着不可替代的作用。以上研究表明,5fc不仅仅作为DNA去甲基化的中间产物,还在多种生物学过程中发挥重要作用,可作为新的疾病标志物。
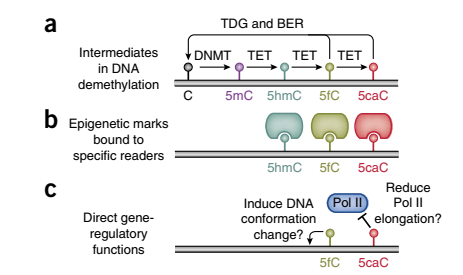
Figure 1:DNA去甲基化过程及各中间产物的功能
数谱生物(原康成生物)为您提供一站式DNA甲酰基胞嘧啶(5fc)修饰测序技术服务,您只需要提供保存完好的组织或细胞标本,我司就可以为您完成从DNA提取、5fc DNA富集、文库构建、高通量测序到数据分析的一整套服务,并提供完整的实验报告。
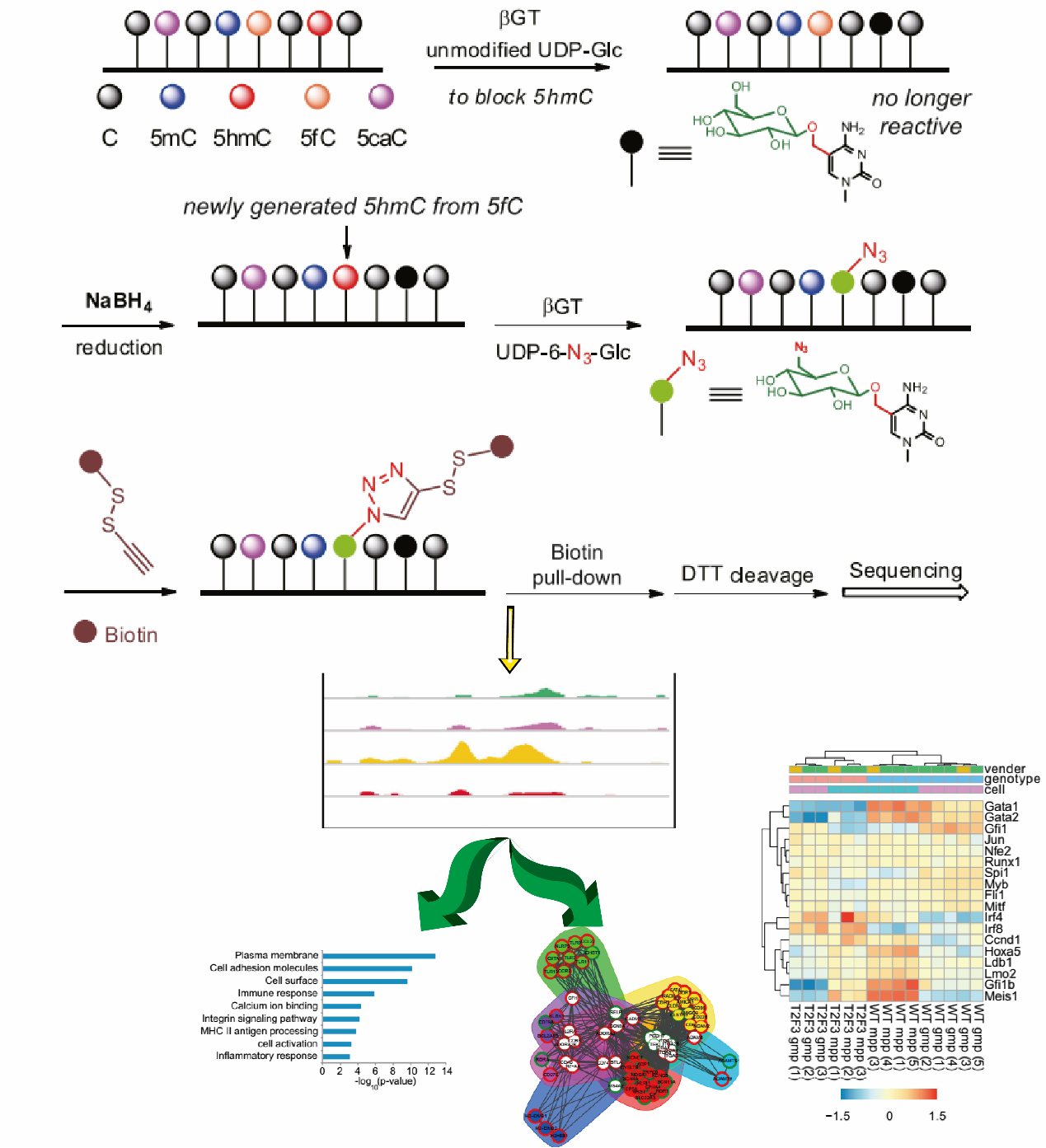
图释:DNA甲酰基胞嘧啶(5fc)修饰测序流程示意图
用普通的葡萄糖在bGT催化作用下将内源的5hmC葡糖基化,然后在NaBH4作用下将5fC还原成5hmC。使用5hmC选择性的化学标记方法(hMe-seal)特异性富集从5fc还原产生的5hmC,进行测序。
高度特异性: 本方法可以从DNA修饰中特异性识别5fC。
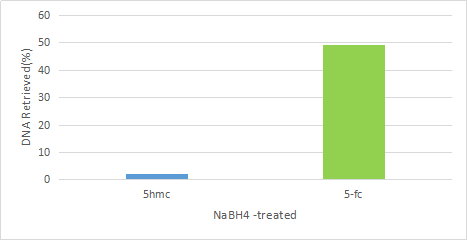
图释:分别对加入了含有2种不同修饰的spike in(分别为5 hmC和5fc)的DNA样品(20 g)进行富集试验,富集完成后进行PCR检测,结果显示该方法具有良好的5fc富集特异性。
无密度偏好性: 本方法富集5fc区域不依赖于DNA修饰的表达水平。
稳定性高:实验操作稳定性高,减少实验操作带来的误差。
灵活度高:能够直接对有基因组信息的任意物种的DNA 5fc进行测序。
精确度高:能够在实际结合位点50个碱基范围内精确定位。
提供文章发表级别的数据可视化图谱
1. peak识别及注释
我司使用MACS2进行peak识别,并根据Ensembl数据库和Ehancer数据库(EnhancerAtlas)的注释信息,对peak进行详细注释。
(1)Peak分类
根据peak顶点与转录本的相对位置关系对peak进行分类,具体分类方法如下。
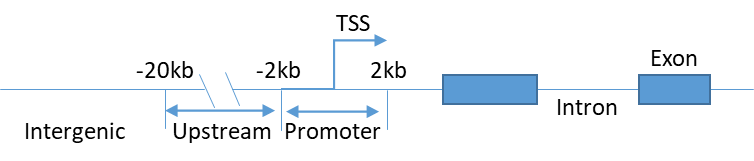
Figure1. peak分类
Table1. 各样本peaks区域

(2)peak统计
根据以上分类,对不同区域进行统计绘制饼状图。
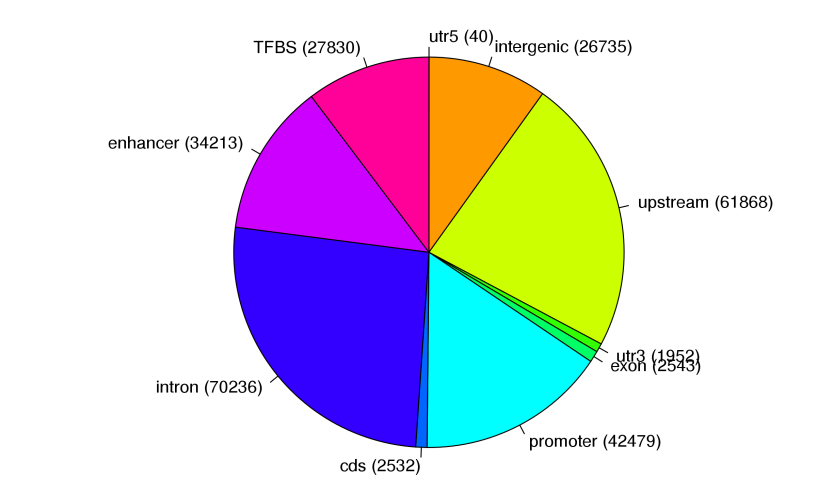
Figure2. peak统计图(TFBS: 转录因子结合位点)
对peaks和fragments在TSS(转录起始位点)及转录本周围的分布情况进行统计并绘图。
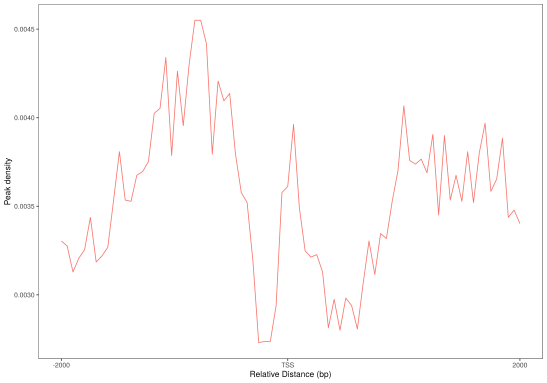
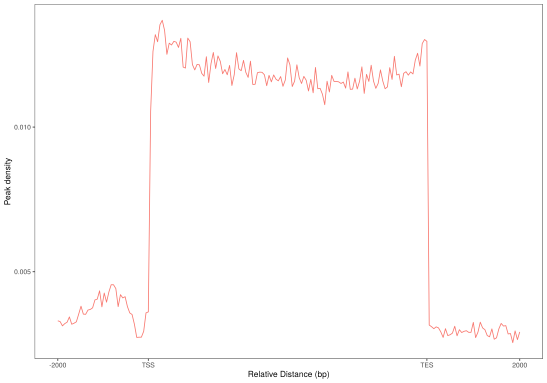
(a)Peaks密度在TSS周围的分布 (b) Peaks密度在转录本周围的分布
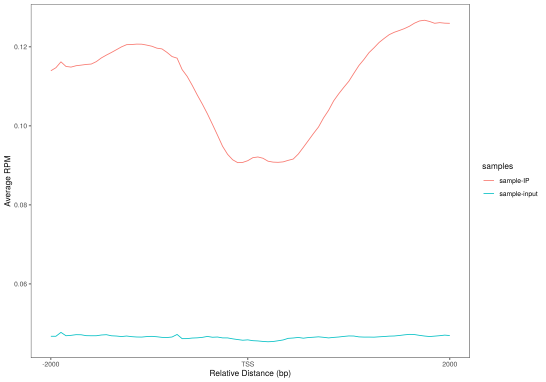
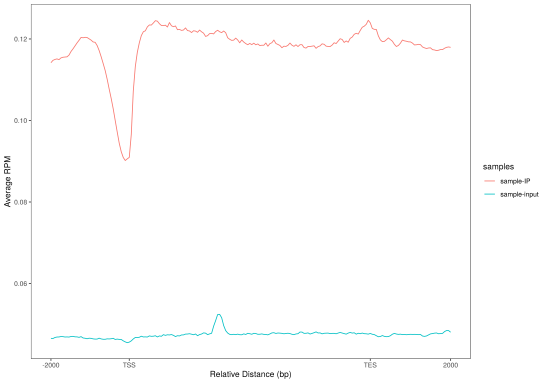
(c)标准化后的reads在TSS周围的分布 (d) 标准化后的reads在转录本周围的分布
Figure3. peak分布情况
修饰密度(peaks/C)用来展示修饰区域在整个染色体上的分布情况;将修饰水平分为低(0%~30%,绿色),中(30%~70%,蓝色),高(70%~100%,红色)三类,在Circos图中进行展示(bin=1Mbp)。
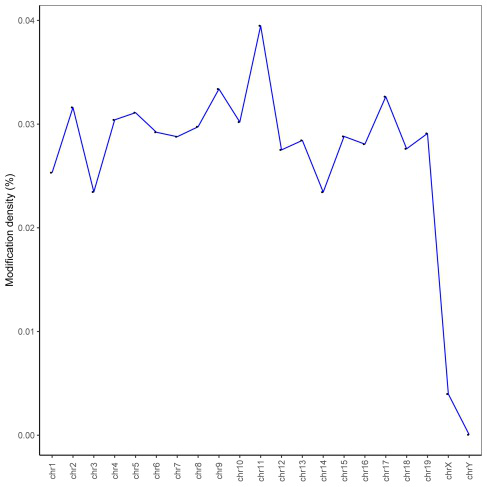
Figure4. 修饰密度在不同染色体上的分布
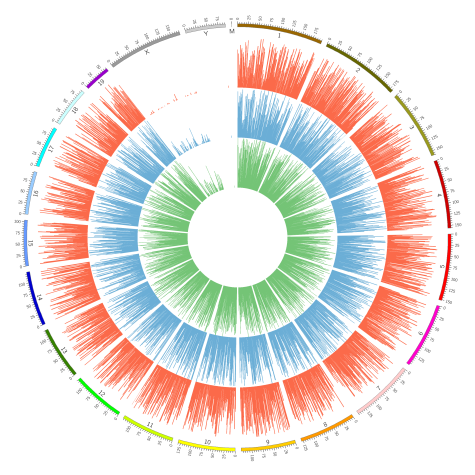
Figure5. 不同修饰水平peaks在染色体上的分布
2. 差异peak分析
康成生物丨数谱生物使用DiffBind进行组间或样本间比较的差异peak分析,并根据Ensembl数据库和Enhancer数据库(EnhancerAtlas)进行差异peak注释。
Table2. 差异peak区域

根据差异peak注释到的不同区域,对区域数量进行统计绘制条形图,并以10kb为单位对peak在染色体上的分布进行统计并绘图。
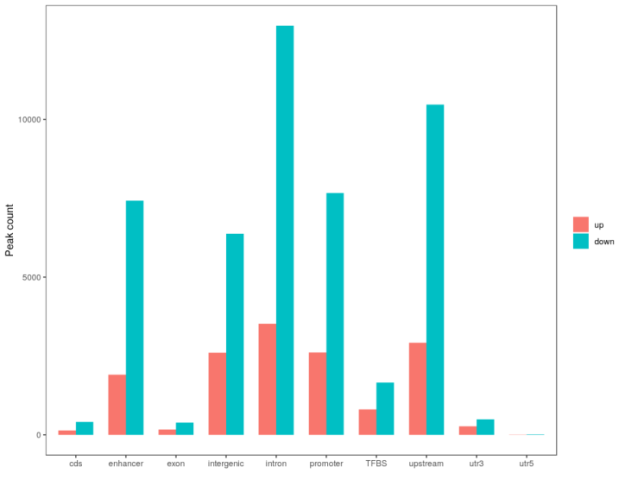

Figure6. 差异peak统计展示 (左):差异peak分布统计图; (右):差异peak染色体分布图
通过各样本在差异peak富集程度的系统聚类分析,可以了解各组样本间的关系。火山图通过-log10P_value和log2Fold_Change两个条件,对比较数据进行筛选,可以直观地展示两组数据之间富集差异的倍数变化及显著性的关系。
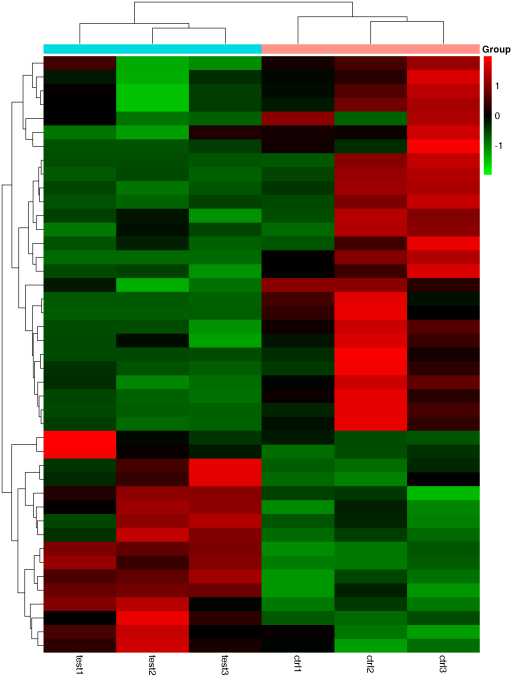
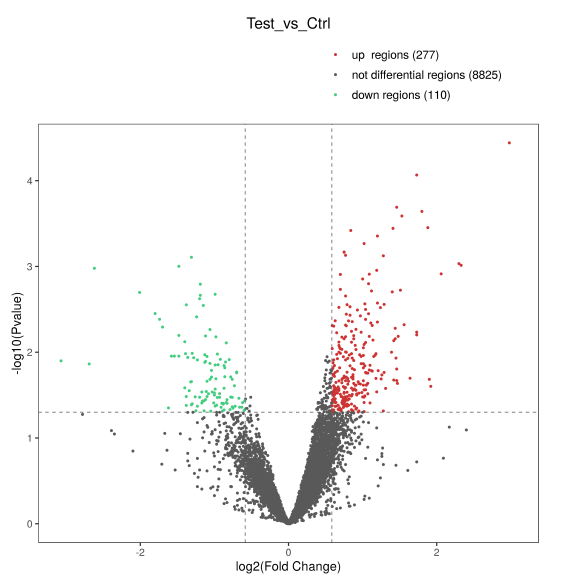
Figure 7. 差异peak聚类图和火山图 (左):聚类图; (右): 火山图
3.差异peak的GO & KEGG富集分析
(1)差异peak的GO富集分析
GeneOntology (GO)是一种基因功能分类条目,有三个子条目,分别描述每个基因的分子功能(MF: Molecular function)、细胞组成(CC: Cellular component)、参与的生物学过程(BP: Biological process)。通常的GO富集分析就是利用统计学算法来找出一组差异表达基因和哪些具体功能条目联系最大,每个GO条目都对应一个统计值P-value来表示显著性,P-value越小表示该GO条目和输入的差异表达基因联系越大,即该组差异表达基因大部分具有该GO条目对应的描述功能。
Table3. GO富集分析结果

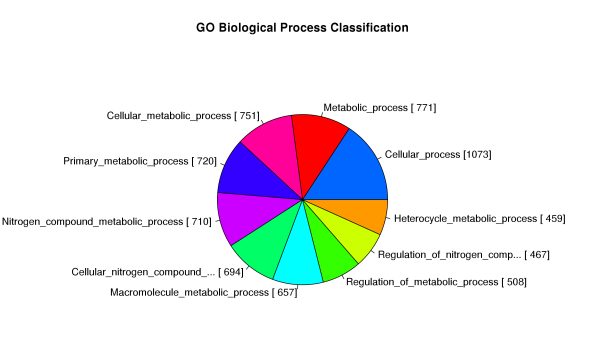
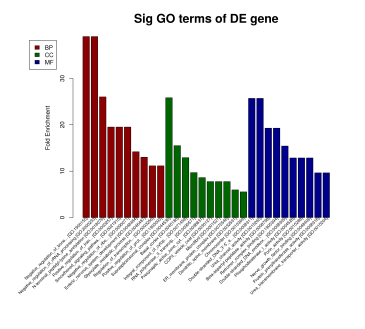
Figure 8. GO富集结果图 (左):各GO条目中出现的基因数;(右)MF、CC、BP前十富集条目柱状图
(2)差异peak的Pathway富集分析
根据KEGG database (Kyoto Encyclopedia of Genes and Genomes)中的生物学通路分类条目,来找出一组差异表达基因和哪些具体的功能条目联系最大。每个Pathway条目都对应一个统计值P-value来表示显著性,P-value越小表示该GO条目和输入的差异表达基因联系越大,即该组差异表达基因P-value越小表示该Pathway条目和输入的差异表达基因联系越大。
Table 4. KEGG富集分析结果

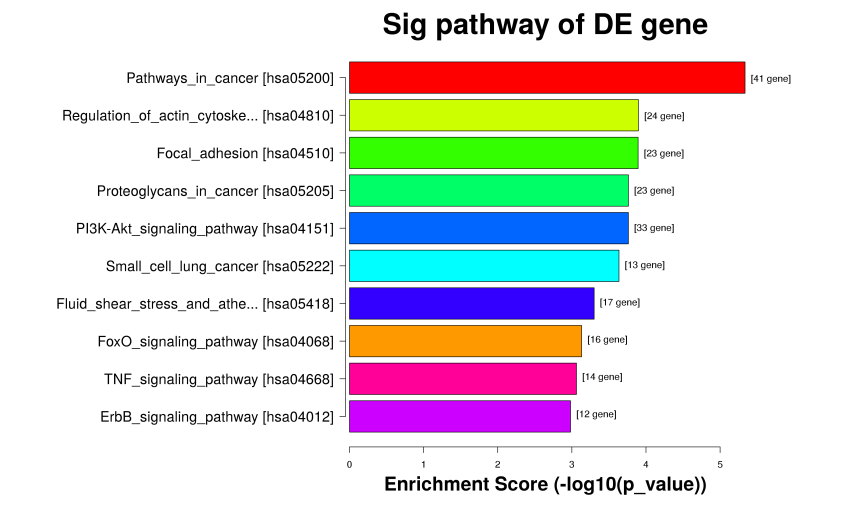
Figure 9. KEGG富集结果图
4. Motif分析
在遗传学中,motif是指一段短的且普遍存在的核苷酸或氨基酸序列。被认为有特定的生物学意义,比如DNA上的蛋白结合位点。当motif出现在基因外显子区域时,其对蛋白结构具有重要作用。我司使用MEME-ChIP(v4.9.1)中的DREME方法(适合于查找短的motif)来检测motif。将得到的motif与转录因子结合位点motif数据库(JASPARCORE2018)内的motif进行比较,从而得到相关motif的更多信息。
Table 5. Motif比较结果



Figure 10. Motif结果图 (左):Motif结果图;(右)左图中序列反向互补图
5. 比对结果可视化
我司将测序数据储存为bedGraph文件,可以在IGV或UCSC基因组浏览器中查看。
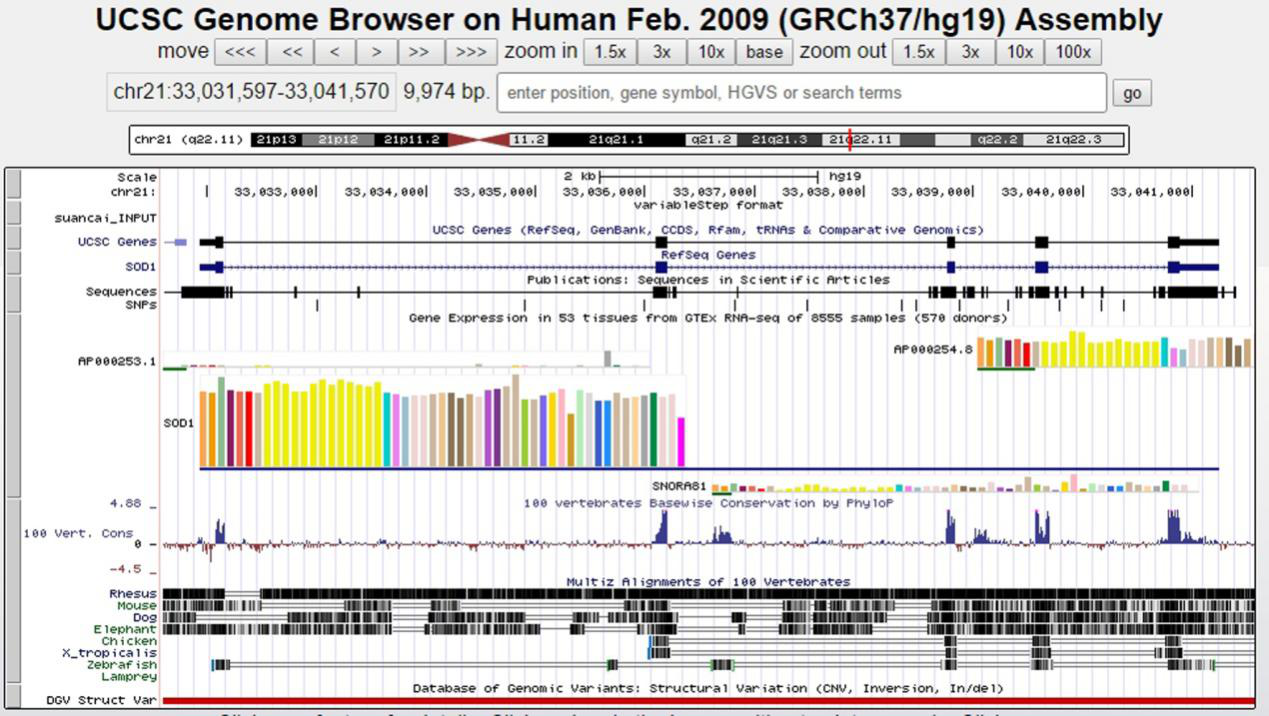
Figure 11. 比对结果可视化


