|
单细胞测序技术服务 靶向单细胞测序(lncRNA&mRNA) 单细胞测序 |
|
蛋白表达定量 DIA定量蛋白质组学 Label free非标定量 |
蛋白修饰定量 N-糖基化蛋白组学 O-GlcNAc修饰蛋白质组学 |
|
Ribo-seq Ribo seq(ribosome profiling) |
核糖体-新生肽链复合物(RNC) RNC联合 circRNA芯片 RNC联合 lncRNA芯片 RNC-seq |
|
NGS测序技术服务 环状DNA测序(eccDNA测序) |
PCR技术服务 环状DNA PCR技术服务 |
在RNA-seq中,RPKM标准化不适用于比较样本间的RNA水平差异
在RNA-seq分析中,RPKM(Reads Per Kilobase of transcript per Million mapped reads)的提出旨在消除文库大小(library size)和基因长度(gene length)对基因表达量定量的影响。
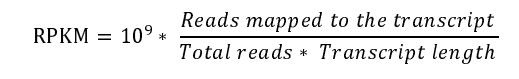
然而,该方法隐含一个关键假设:即所有样本的总RNA含量相同。RPKM计算的本质是描述单个样本内某转录本的相对丰度(relative abundance),而非其绝对表达量。当样本间整体RNA组成存在差异时,RPKM的标准化结果会产生系统性偏差。例如,即使转录本Y的真实表达水平保持不变,其RPKM值也可能由于样本中其他转录本表达的变化而显得更高或更低[1]。
RNA-Seq无法可靠地定量低丰度转录本
由于 RNA-Seq 对测序深度的依赖性和读数的固有变异性,RNA-Seq 在准确定量低丰度转录本方面存在困难。这种变异性会导致检测在低丰度 RNA 时,因为测序的随机性导致读数不足,从而无法进行准确量化。对于低丰度 RNA,有限的读数深度所导致的泊松采样噪声是 RNA-Seq 的主要误差来源[2]。因此,RNA-Seq 无法可靠地测量表达量低的转录本,需要对其进行富集[2]。
使用三个技术重复数据集(每个数据集包含 3.31 亿个 50bp 读数),对基因表达水平与测量精度之间的关系进行了研究和表征[3]。如图1所示,基因表达水平越高,测量越精确。相反,基因表达水平越低,测量的相对误差就越大。
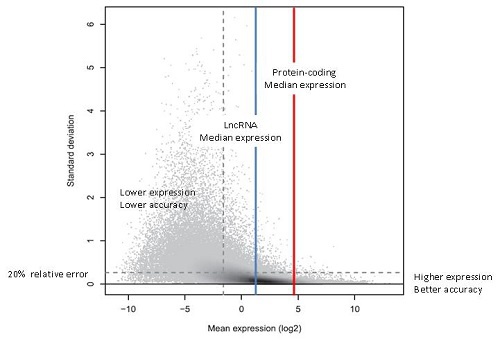
图1. 标准偏差与表达水平。该图显示了三次技术重复测量的变化(标准偏差,Y 轴),每个可辨认的点代表一个转录本。在阴影区域,灰度代表密度,深色阴影表示密度较高。平均表达水平较低的转录本(x 轴)的标准偏差一般较大。表达较强的转录本通常可以得到可靠的测量,相对误差不超过 20%。有趣的是,在所有转录本靶标中,只有 41% 的靶标能以这样的精度进行测量(水平虚线以下)。另一方面,在 41% 表达最强的转录本(垂直虚线右侧)中,有 84% 可以得到可靠的测量(水平虚线以下)。这反映在图中就是右侧(深色阴影)的目标物密度很高,大部分都在水平线以下,而垂直虚线左侧的情况并非如此[3]。
增加测序深度对低丰度转录本的准确性提升有限
为了更准确地测量表达水平,可以增加 RNA-seq 的测序深度。一般来说,100M reads足以检测大多数表达基因和转录本,但要准确定量大多数(即 72%)基因的表达水平,则需要 500M reads [4]。然而,增加测序深度只会对高表达的转录本的准确性产生显著影响,表达量低的转录本的准确性则相对不会随着测序深度的增加而改变,这一点在不同丰度水平的 PHB、CD74 和 BRD4 转录本异构体检测的案例研究中可以看到[4]。
总体而言,RNA-seq 测序深度的无限增加不会导致可靠定量的低丰度转录本数量的无限增加。测序深度的增加会导致收益递减,最终达到最大值(图 2)[3]。在 RNA-Seq 中,7% 的高丰度转录本占据了所有reads比对的 75% 以上。大部分增加的reads都浪费在了少数丰富的转录本上,比如管家基因。这意味着某些低丰度 RNA 的表达水平在实际测序深度下无法精确测量。
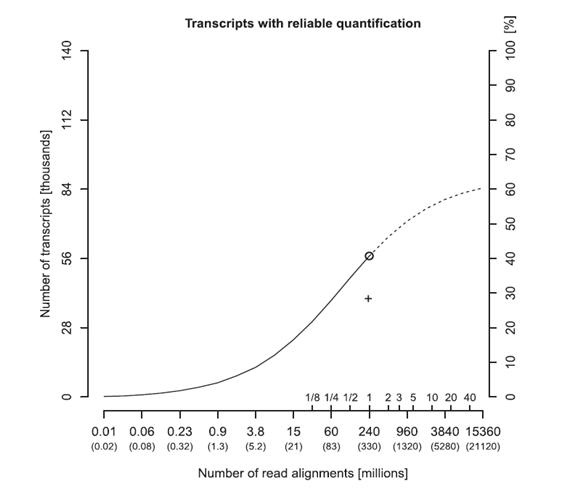
图2. 可靠定量的转录本(相对误差不超过 20%)与测序深度的关系。横轴括号中给出了总reads数。附加的内部刻度线表示测序运行的次数。右侧的备用 Y轴显示了可靠测量的所有已知转录本的百分比。圆圈符号表示整个测序过程中的最大读数(331M reads)。根据拟合的 Sigmoid 曲线推断,即使达到 10000M reads,最大值也只有 60%。
芯片更适合低丰度 RNA 分析
芯片通过其序列特异性探针来检测 RNA。对于特定转录本而言,即使存在高丰度的不相关序列,也不会与探针结合,对检测低表达转录本几乎没有影响。然而,在 RNA-Seq 中,绝大部分测序reads都被高丰度 RNA(如管家基因)占据,低丰度 RNA 的覆盖程度很低,导致灵敏度和可靠性较低。因此,芯片对低丰度 RNA 更具检测优势。例如,芯片常规可检测 7000~12,000 个 LncRNA,而 RNA-seq 需要超过120M reads才能检测 1000~4000 个 lncRNA [5]。在一项临床研究中,选择芯片而不是 RNA-seq,主要是考虑到芯片的灵敏度更高[6]。
参考文献
1.Zhao S et al: (2020) Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. RNA 2020, 26(8):903-909
2. Jiang L et al: Synthetic spike-in standards for RNA-seq experiments. Genome Res 2011, 21(9):1543-1551.[PMID: 21816910]
3. Labaj PP et al: Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics 2011, 27(13):i383-391.[PMID: 21685096]
4. Toung JM, Morley M, Li M, Cheung VG: RNA-sequence analysis of human B-cells. Genome Res 2011, 21(6):991-998.[PMID: 21536721]
5. Kretz M et al: Suppression of progenitor differentiation requires the long noncoding RNA ANCR. Genes Dev 2012, 26(4):338-343.[PMID: 22302877]
6. Xu W et al: Human transcriptome array for high-throughput clinical studies. Proc Natl Acad Sci U S A 2011, 108(9):3707-3712.[PMID: 21317363]


