|
单细胞测序技术服务 靶向单细胞测序(lncRNA&mRNA) 单细胞测序 |
|
蛋白表达定量 DIA定量蛋白质组学 Label free非标定量 |
蛋白修饰定量 N-糖基化蛋白组学 O-GlcNAc修饰蛋白质组学 |
|
Ribo-seq Ribo seq(ribosome profiling) |
核糖体-新生肽链复合物(RNC) RNC联合 circRNA芯片 RNC联合 lncRNA芯片 RNC-seq |
|
NGS测序技术服务 环状DNA测序(eccDNA测序) |
PCR技术服务 环状DNA PCR技术服务 |
lncRNA和mRNA分子结构相似,因此高通量筛选技术可以同时检测这两种类型的RNA。然而lncRNA也具有显著区别于mRNA分子的一些独特特征,这就导致lncRNA的表达谱筛选存在一定的困难,尤其是在分析平台的选择上。 Arraystar的 lncRNA微阵列芯片是专门为克服这些挑战而设计,与NGS(新一代测序平台)相比具有不可替代的优势。
要素1——可以精确检测lncRNA的差异表达
GENCODE的统计结果分析表明,lncRNA的平均表达水平只有mRNA的1/10(下图左)[1]。甚至多数lncRNA在细胞中只有几个拷贝水平的表达,例如lncRNA ANRIL在细胞中只有1-2个拷贝,然而相比之下mRNA FOXF1有成百上千个拷贝(下图中)。另外,lncRNA的表达具有高度的组织特异性和时序特异性,以海马组织为例,不同的lncRNAs在不同的细胞类型和特定组织结构中才会表达(下图右)。尽管少数的lncRNA在特定的细胞中属于高丰度转录本,但是这种高丰度会被稀释在从整个组织块提取的总RNA中。这也是导致lncRNA丰度低的另一重要因素。
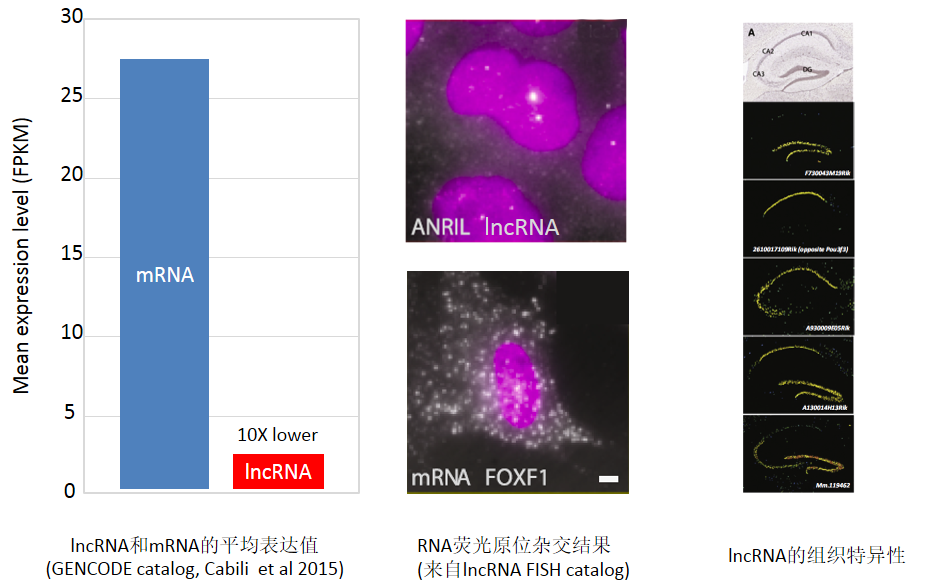
图释:(左) lncRNA和mRNA的表达值统计结果展示图,(中) lncRNA ANRIL与mRNA FOXF1的FISH结果,(右)海马组织中lncRNA的高度组织特异性表达结果展示
尽管细胞中lncRNA的丰度比较低,但是我们也不能忽略对它的研究。毕竟lncRNA和mRNA不一样,它是在transcript水平发挥功能;即使lncRNA有极低的拷贝数,同样可以发挥重要的调控作用。低丰度的lncRNA可以通过多种机制调控临近基因的表达,比如招募染色质重塑复合物来改变染色质的激活状态、招募剪切体来促进转录的起始或者作为eRNA来激活转录等等(图2)。与弥散型分布发挥trans(反式)功能的lncRNA相比,这种cis调控方式不需要lncRNA具有很高的表达水平就可以发挥效应,例如lncRNA-HOTTIP在细胞中只有1-2个拷贝,依然可以招募组蛋白修饰复合物WDR5/MLL发挥cis作用,从而调控临近的HOX基因簇;此外,eRNA(增强子类型的lncRNA)也是在低丰度时发挥的cis调控功能来激活临近基因的表达。
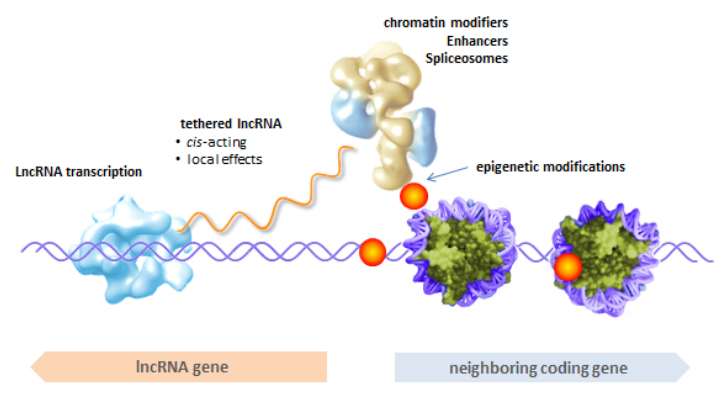
图释:低拷贝的lncRNA通过锚定在转录位点附近来cis调控临近基因的表达
准确定量lncRNA的丰度对于揭示lncRNA在较低水平下如何发挥调控功能是至关重要的。目前测序对于低丰度的lncRNA定量及差异分析存在一定的问题,如:
(1)测序对lncRNA检测的错误率高。测序reads属于count数据,其错误率分布理论上遵循泊松模型,而实际测序过程中,错误率会更高,其分布遵循负二项超离散模型,随着RNA丰度的降低,测序的错误率迅速增加,要保证错误率小于20%,测序count需>100(下图)[3]。

图释:测序错误率与RNA表达丰度关系
(2)测序只能精确定量约4.7%的lncRNA。测序存在高丰度RNA检测偏好,表达丰度最高的前1%的管家基因mRNA占据了测序数据的40%,低丰度的lncRNA的测序覆盖度很小,并且目前数据库收录和注释的lncRNA不全面,导致与数据库进行比对时很多lncRNA reads无法比对上而被忽略掉(下图左)。
利用RNA sequins(已知序列和浓度梯度的一系列外源RNA)作为spike in control,以RNA sequins的观测值与实际值的线性相关程度作为参考评估内源RNA的表达准确性,并用LoQ(Limit of Quantification)作为阈值来反应能准确定量的最小RNA表达水平(下图右),结果发现当FPKM<3.13时,RNA观测值与实际值不呈线性关系并波动剧烈,不能被精确定量,mRNA中只有27.4%丰度满足精确定量要求,lncRNA则只有约4.7% [4]。
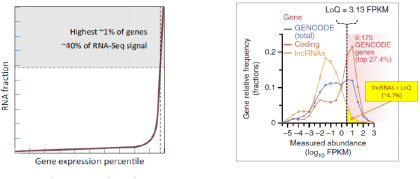
图释:(左)不同表达丰度RNA的测序覆盖度 (右)可被精确定量的lncRNA丰度及占比
(3)测序能进行准确差异表达分析的lncRNA远小于4.7%。利用不同比例梯度的RNA sequins混合物作为spike in加入实验组和对照组,随后进行测序,评估内源RNA的差异表达显著性(下图左),结果表明差异倍数越大且表达丰度越高,差异结果越显著(P值越小)。利用LODR(Limit of Detection of Ratio)作为阈值评估差异结果显著(P<0.05, FC>2)需要的最小RNA表达水平,发现极少数lncRNA满足准确分析显著差异的表达丰度(下图右),数量远远低于测序能准确定量的lncRNA(约4.7%)[4]。
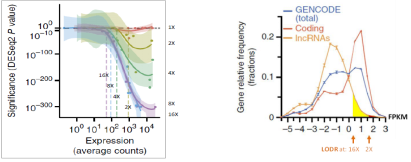
图释 :可准确分析表达差异显著性的lncRNA丰度及差异倍数
而微阵列芯片是依靠固定在芯片上的寡核苷酸探针去捕获并富集特异性靶序列,然后检测相应的荧光信号值。因此,与RNA-seq相比,微阵列的准确性受低丰度转录本的影响较小,2010年PANS发表的一篇文章同样也佐证了该观点[5]。
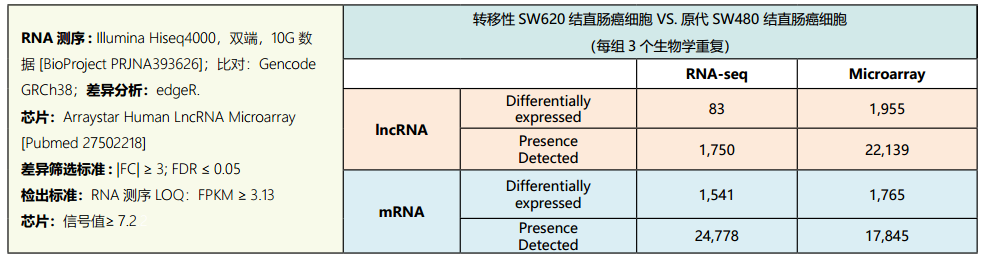
下表比较了测序和芯片对相同细胞进行lncRNA检测和差异分析的结果,由结果可以看出,芯片较测序能检测更多的差异表达lncRNA:
要素2——可以区分和检测lncRNA复杂且具有重要功能的转录异构体
由于缺乏像mRNA那样具有开放阅读框的限制,lncRNA通常会被转录并剪切成各种转录异构体,并且这些异构体可能具有不同甚至相反的生物学功能。基因MEG3可以转录产生12个异构体,但是这12个异构体对于靶基因P53的激活和肿瘤的抑制程度各有差异[6]。另外,基因BCL2L1转录生成的lncRNA BCL-XL、BCL-XS和ENST具有相反的生物学调控功能[7]。由于RNA-seq存在reads不完全覆盖转录本、以及从头拼接转录本的复杂性和不可靠性,导致RNA-seq对于lncRNA的定量不准确。然而,Arraystar lncRNA芯片是基于“一个转录本一个探针”的设计原理进行设计,可以准确检测不同异构体的表达。

图释:MEG3转录的不同异构体对于p53的激活程度各有差异
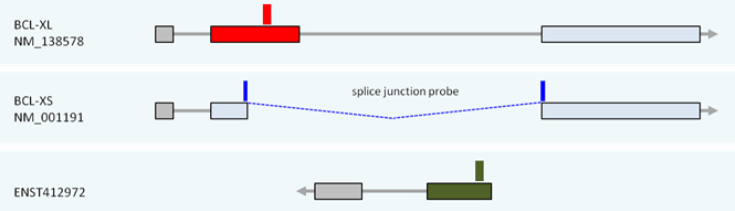
图释:Arraystar lncRNA芯片通过设计独特探针来检测BCL的不同转录异构体
要素3——提供lncRNA详细的注释和分析
尽管lncRNA的研究越来越热,但是由于缺乏开放阅读框可以参考,所以和mRNA相比,大多数lncRNA仍然是注释信息匮乏,功能不明确。为了有效分析lncRNA的表达水平和相应的生物学功能,Arraystar lncRNA芯片标准软件包集成了丰富的,详细的,系统的lncRNA注释和分析,以及该领域的最新研究进展,可以为用户提供一站式芯片服务。最新升级的Arraystar lncRNA芯片更是新增lncRNA的序列完整性、表观基因组信息、是否有DNA酶I超敏位点(DHS)、短肽编码潜能及亚细胞定位等详细注释,帮助研究者更快锁定研究目标。
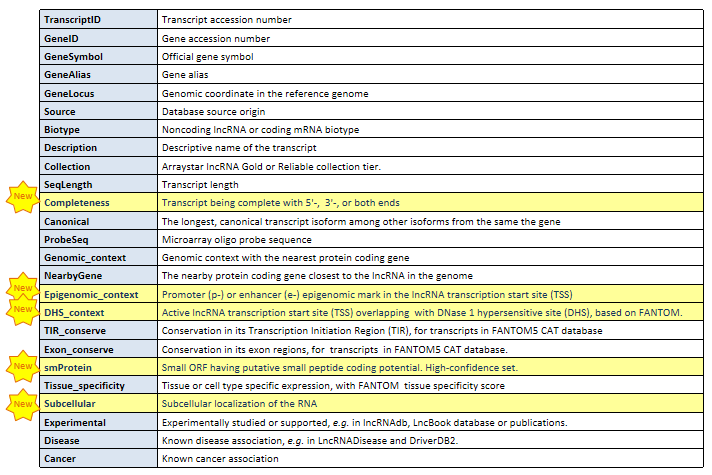
图释:Arraystar lncRNA芯片结果对于lncRNA进行系统和详细的注释和分类
参考文献
[1]Cabili M.N. et al. (2015) "Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution." Genome Biol. 16:20 [PMID: 25630241].
[2]Wang K.C. et al. (2011) "A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression." Nature 472(7341):120-4 [PMID: 21423168]
[3]Engreitz J.M. et al. (2016) "Local regulation of gene expression by lncRNA promoters, transcription and splicing." Nature 539(7629):452-455 [PMID: 27783602]
[4]Xu W. et al. (2011) "Human transcriptome array for high-throughput clinical studies." Proc. Natl. Acad. Sci. U.S.A. [PMID: 21317363]
[5]Anders, S. et al .(2010). Differential expression analysis for sequence count data. Genome biology, 11(10), R106. [PMID: 20979621]
[6]Hardwick S. A. et al . (2016). Spliced synthetic genes as internal controls in RNA sequencing experiments. Nature methods, 13(9), 792. [PMID: 27502218 ]
[7]Zhang X. et al. (2010) "Maternally expressed gene 3 (MEG3) noncoding ribonucleic acid: isoform structure, expression, and functions." Endocrinology 151(3):939-47 [PMID: 20032057]
[8]Boise L.H. et al. (1993) "bcl-x, a bcl-2-related gene that functions as a dominant regulator of apoptotic cell death." Cell 74(4):597-608 [PMID: 8358789]
[9]Hon C.C. et al. (2017) "An atlas of human long non-coding RNAs with accurate 5' ends." Nature 543(7644):199-204 [PMID: 28241135]


