|
单细胞测序技术服务 靶向单细胞测序(lncRNA&mRNA) 单细胞测序 |
|
蛋白表达定量 DIA定量蛋白质组学 Label free非标定量 |
蛋白修饰定量 N-糖基化蛋白组学 O-GlcNAc修饰蛋白质组学 |
|
Ribo-seq Ribo seq(ribosome profiling) |
核糖体-新生肽链复合物(RNC) RNC联合 circRNA芯片 RNC联合 lncRNA芯片 RNC-seq |
|
NGS测序技术服务 环状DNA测序(eccDNA测序) |
PCR技术服务 环状DNA PCR技术服务 |
在MYC驱动的肿瘤增殖过程中,MYC结合新生RNA并限制R-loop累积和固有免疫通路激活,成为了理解肿瘤生长和免疫逃逸的新颖作用机制。2026年发表于《Cell》(IF=42.5)的研究揭示:利用eCLIP-seq、ChIP-seq、DRIPc-seq等联合分析发现,在应激条件下,癌蛋白MYC从DNA结合状态转变为RNA结合状态,并围绕R-loop形成多聚体,聚集RNA降解因子,限制异常R-loop积累,阻止其泄漏至细胞质,抑制TLR3-pTBK1固有免疫信号通路,促进免疫逃逸和肿瘤生长。
背景介绍:MYC的双重身份
MYC蛋白是多种肿瘤的致癌驱动因子,在人类肿瘤中表达升高。其经典作用是作为转录因子,与MAX形成复合物结合DNA,驱动肿瘤细胞生长和增殖。然而,并非所有MYC的致癌效应都能通过基因表达变化来解释。例如,MYC会建立“冷肿瘤”免疫微环境,使肿瘤逃避免疫监视。
机制上,MYC抑制TANK结合激酶1(TBK1)的活性,后者可激活NF-κB和干扰素表达。MYC缺失细胞中TBK1的激活依赖于Toll样受体3(TLR3),这是一种识别双链RNA及R-loop来源RNA-DNA杂交体的模式识别受体。但MYC如何影响dsRNA或R-loop积累尚不清楚。
值得注意的是,同为MYC家族的MYCN已被证实能结合RNA,作为核外泌体靶向(NEXT)复合物的一部分,促进内含子RNA降解。这提示MYC也可能具备类似的RNA结合功能。
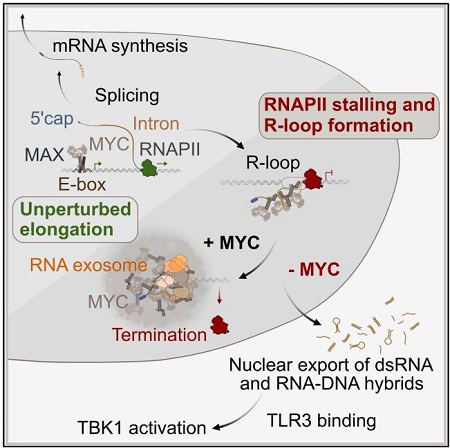
图1:MYC结合RNA影响R-loop积累与固有免疫通路激活的机制图。
实验思路图
实验结果展示
核心发现:MYC结合新生RNA,从DNA“跳槽”到RNA
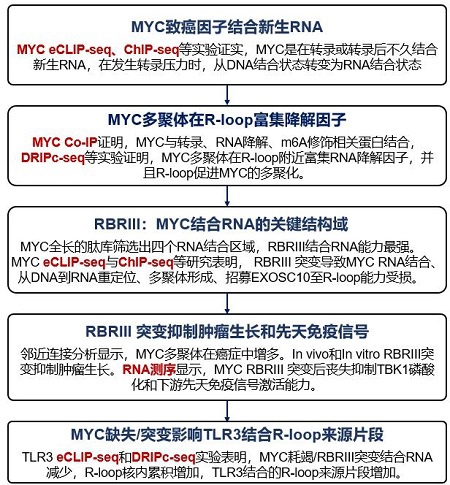
研究团队首先通过eCLIP-seq实验证实,MYC能够结合RNA,且约70%的结合位点位于内含子RNA中,约10%邻近剪接位点,表明MYC主要结合新生RNA。进一步实验发现,使用CDK9抑制剂NVP-2阻断新生RNA合成后,MYC的RNA结合显著降低,证明MYC是在转录过程中或转录后不久结合新生RNA的(图1)。
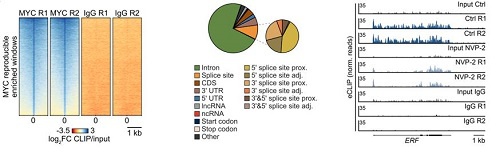
图2:MYC与新生RNA结合
那么,MYC在DNA和RNA之间的结合状态是否会动态变化?eCLIP-seq和ChIP-seq等研究发现,当FACT组蛋白伴侣(调控转录延伸)或剪接体SF3B1(调控RNA剪接)受到抑制时,内含子RNA积累,MYC从DNA结合状态转变为RNA结合状态。类似地,敲低核外切体核心亚基EXOSC5(负责降解内含子RNA)也导致同样的结果。
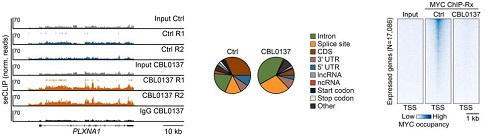
图3: MYC的RNA结合和DNA结合状态变化
MYC多聚体:一个围绕R-loop的RNA降解中心
既然MYC多聚体是围绕新生RNA形成的,那么它很可能在RNA代谢中发挥作用。Co-IP免疫共沉淀实验显示,MYC不仅与MAX、RNAPII等转录相关蛋白结合,还与EXOSC10(外切体催化亚基)、ZCCHC8(NEXT复合物支架) 等RNA降解因子,以及m⁶A修饰相关蛋白相互作用。
更重要的是,在转录压力下,dsRNA和R-loop均富集在MYC多聚体内部。DRIPc-seq显示,CBL0137处理细胞中鉴定出的R-loop与MYC RNA结合位点重叠。去除R-loop可降低MYC多聚化。这表明MYC多聚体在R-loop和dsRNA附近富集RNA降解因子,形成一个“RNA降解中心”。
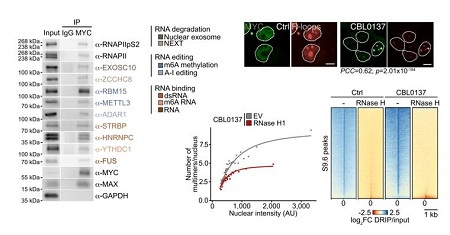
图4:MYC多聚体是围绕R-loop的RNA降解中心
RBRIII:MYC结合RNA的关键结构域
通过覆盖MYC全长的肽库筛选,研究鉴定出四个RNA结合区域(RBR I–IV),其中RBRIII结合RNA能力最强。构建RBRIII六位点丙氨酸突变体(RBRIII MUT MYC)后,体外结合实验证实其RNA结合能力丧失。
MYC eCLIP-seq与ChIP-seq等研究表明,与野生型MYC相比,RBRIII MUT MYC在细胞中表现出以下缺陷:结合新生RNA能力受损;从DNA到RNA的重新定位受阻;多聚体形成能力下降;与EXOSC10共定位及将其招募到R-loop的能力显著降低。
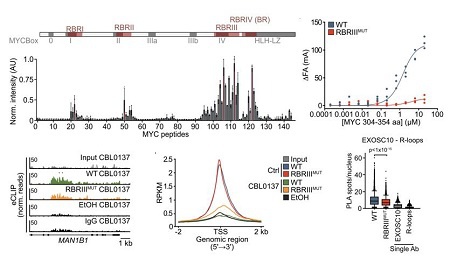
图5:MYC结合RNA的RBRIII突变影响RNA结合、DNA到RNA重定位、与EXOSC10共定位及将其招募到R-loop的能力
RBRIII对肿瘤生长至关重要,且抑制先天免疫信号
邻近连接分析显示,MYC多聚体在正常组织中几乎检测不到,但在人类结肠癌和胰腺癌组织中水平显著升高。
在胰腺KPC细胞模型中,野生型MYC显著促进细胞增殖,RBRIII MUT MYC和shMYC增殖减弱。在体内实验中,表达野生型MYC的肿瘤28天内平均体积增加24倍,而表达RBRIII MUT MYC的肿瘤体积减少94%。这说明RBRIII在体外培养中并非必需,但对体内肿瘤维持至关重要。
RNA测序显示,野生型MYC能够抑制NF-κB/RelA、视黄酸受体、干扰素调节因子Irf1等先天免疫相关转录因子驱动的基因,而RBRIII MUT MYC则不能。此外,RBRIII MUT MYC无法抑制TBK1磷酸化激活及下游信号。
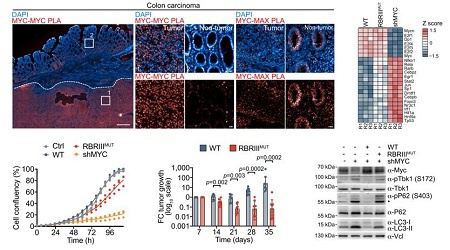
图6:RBRIII对肿瘤生长至关重要,且抑制TBK1激活及先天免疫信号
RBRIII介导R-loop结合,抑制TLR3 DNA/RNA杂交体积累
MYC耗竭后TBK1的活化依赖于TLR3,该受体能结合dsRNA和R-loop来源的DNA/RNA杂交体。TLR3 eCLIP-seq和DRIPc-seq实验表明,MYC耗竭后TLR3结合的RNA数量增加,R-loop在TLR3上的积累也增加。
R-loop全基因组图谱分析鉴定出4,912个R-loop区域,其中501个与MYC结合的RNA重合,且大多数位于内含子中。表达RBRIII MUT MYC的细胞中,这些R-loop的量显著高于野生型,而MYC与这些R-loop的结合量则显著降低。
单基因可视化显示,RBRIII MUT MYC或MYC耗竭组中,MYC结合的RNA减少;而DRIPc-seq显示R-loop积累增加;TLR3eCLIP-seq显示这些R-loop中的RNA在MYC耗竭后会被TLR3结合。
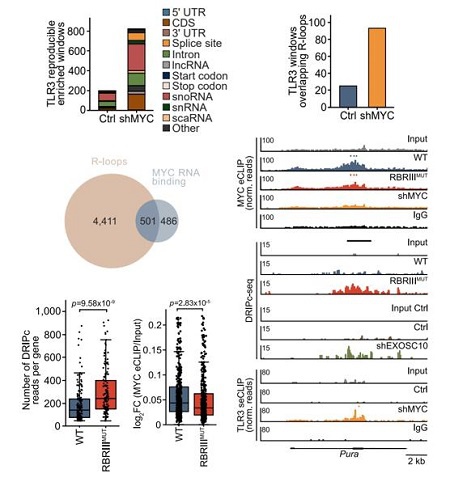
图7:MYC通过结合RNA,抑制RNA-DNA杂交体在TLR3上的积累
总结与展望
本研究颠覆了MYC仅作为经典转录因子的传统认知,揭示了其作为多价RNA结合蛋白的新身份。核心机制是:利用eCLIP-seq、ChIP-seq、DRIPc-seq等联合分析发现,当肿瘤细胞面临转录压力时,MYC从DNA结合状态切换至RNA结合状态,通过RBRIII结构域介导多聚化,将核外切体等RNA降解因子富集于R-loop和dsRNA周围,清除这些免疫原性核酸结构,抑制TLR3-TBK1先天免疫通路,最终帮助肿瘤实现免疫逃逸。
这一发现首次将MYC的RNA结合活性、多聚化能力与肿瘤免疫微环境重塑直接联系起来,为理解MYC致癌机制提供了全新维度。未来可针对MYC的RBRIII结构域开发小分子或多肽抑制剂,阻断其RNA结合与多聚化能力,从而“唤醒”肿瘤内的先天免疫应答,增强免疫治疗疗效。
研究信息: 该研究由德国维尔茨堡大学等单位完成,发表于《Cell》(2026年1月),DOI: 10.1016/j.cell.2025.12.019。
康成生物|数谱生物能提供的相关技术服务


