摘要
染色质发生改变与肿瘤的发生、发展的各个阶段密切相关。特征性地表现在表观遗传调控引起基因转录沉默,转录沉默与调控细胞重要功能基因的启动子甲基化相关。最近发现在肿瘤发生早期,表观遗传的改变首先引起肿瘤细胞的信号传导途径发生改变,由于信号通路改变促使细胞信号途径中其他基因突变,由此细胞获得选择优势促进肿瘤的发生和发展。逆转表观遗传调控的基因沉默有可能应用于肿瘤的干预和治疗。
通常认为肿瘤是由渐进性的遗传异常驱动的一种疾病,这些遗传异常包括肿瘤抑制基因与致癌基因突变和染色体异常。然而,肿瘤显然也是由表观遗传改变引起的一种疾病,表观遗传是指不引起基因序列改变的可遗传改变,这种改变影响基因表达的变化。表观遗传的变化包括DNA甲基化的丢失、获得,以及组蛋白修饰的改变。目前发现越来越多的肿瘤抑制基因和潜在的肿瘤抑制基因在转录水平上被抑制,这种抑制与DNA甲基化水平改变相关。通过这种沉默机制,肿瘤细胞中的肿瘤抑制基因的表达降低或者完全消除,这可能也是遗传变异的一种替代机制。
人们越来越清楚的认识到表观遗传改变对于肿瘤发生的重要性,该领域的研究已经从研究经典的肿瘤抑制基因沉默的影响发展到根据启动子区域是否高甲基化来寻找可能的肿瘤抑制基因,甚至已经开始研究肿瘤细胞整个基因组或者基因组上某一特定区域的甲基化和染色质状态的改变。最近的研究表明在肿瘤形成的早期,表观遗传的改变会引起癌前细胞的扩增。这部分细胞最先是发生表观遗传的改变,表观遗传的改变决定了随后的遗传改变,遗传改变则促使这些克隆恶变。基因组的甲基化模式已经用于肿瘤分型、肿瘤危险评估、早期检测、监控和诊断的标志以及评估肿瘤易感性或治疗后反应的指标。
概要
● 表观遗传调控的基因沉默是癌症中基因功能缺失的一个重要的机制,这种基因沉默与启动子甲基化的异常及转录抑制相关。
● 表观遗传调控的基因沉默发生在人类肿瘤发生的早期(发生转移前的病灶),干扰或激活关键的信号通路。
● 肿瘤早期发生的基因沉默事件,通过使关键的细胞信号通路改变,促使细胞发生异常的早期克隆性扩增。
● 早期的基因沉默事件有可能诱导细胞依赖某一癌症相关细胞信号通路,并使得遗传变异在统一信号通路中累积,从而促使肿瘤的发生发展。
沉默的基因
肿瘤细胞基因组呈现出整体甲基化水平降低和基因启动子特异性的甲基化水平升高的状态。最近的一些综述针对甲基化水平升高如何调控转录水平上基因沉默的机制进行了讨论。目前还不清楚肿瘤恶化过程中基因组整体甲基化水平为何降低,但是整体甲基化水平降低可能会引起基因组不稳定、染色质结构改变以及一些基因表达上升。目前的研究主要集中在启动子的甲基化和随后的基因表达降低,这些变化对于肿瘤生物学的影响变得越来越明显。在肿瘤细胞中许多基因是由于表观遗传修饰而丧失功能,而不是通过遗传缺失。
在研究人结肠癌细胞株HCT116中发现,遗传改变和表观遗传调控的基因沉默共同决定了肿瘤细胞的表型。HCT116细胞中一些基因发生了突变,肿瘤抑制基因失活而致癌基因被激活,这就导致了关键的信号通路和细胞的功能发生紊乱(表1)。这些基因突变包括MLH1和p16蛋白的基因,其表达产物分别调控错配修复和cyclin D-RB1控制的细胞周期途径。发生突变的基因还有转化生长因子βⅡ受体(TGFβ2R),该基因的突变导致影响细胞分化的信号通路失调。并且,HCT116细胞中还有一些基因因突变而激活,比如β-catenin,最终导致Wnt信号的持续激活和细胞增殖。
然而,除了这些特异性的基因突变,在这些细胞中还发现至少14个表观遗传调控沉默的基因。当用DNA去甲基化药物处理细胞或者沉默编码DNA甲基转移酶的基因后,受表观遗传调控沉默的基因又能够重新被激活。这些基因重新激活则导致细胞表型发生改变,包括减少增殖、诱导凋亡(表1)。此外,在HCT116细胞中MLH1和CDKN2A(编码p16蛋白)基因同时发生表观遗传异常和遗传异常。当细胞中这些基因的一个等位基因发生突变,那么野生型的等位基因则由于甲基化水平升高而沉默。因此,在肿瘤细胞中,遗传和表观遗传的改变共同阻止一些功能性的基因表达。
HCT116细胞中表观遗传和遗传改变共同发生作用的情况还出现在Wnt信号途径中。在Wnt信号通路中frizzled相关基因家族的四个成员(SFRP1、SFRP2、SFRP3和SFRP4)编码分泌型Wnt拮抗蛋白,在HCT116细胞中,这四个基因被表观遗传调控而沉默,导致Wnt信号异常激活,这一现象甚至在β-catenin发生激活性突变的细胞中发生。另外,编码转录因子GATA4和GATA5的基因以及它们下游的目标基因TFF1、TFF2、TFF3和inhibin-α发生沉默抑制内胚层来源的上皮细胞的成熟。此外,HCT116细胞中TIMP3(组织金属蛋白酶抑制因子3)发生沉默,其表达的蛋白功能缺失有可能增加细胞的迁移能力。
研究肿瘤细胞的表观遗传谱和遗传谱一样重要,它有助于我们理解不同的肿瘤细胞为何进化出不同表型。目前已经提出在结肠癌以及其他一些类型的癌症中细胞呈高甲基化的表型,其中一些特异的基因通过启动子的甲基化水平升高而发生沉默。这些改变使得细胞获得选择优势从而促使肿瘤生长和恶化。这一过程中涉及的机理有待进一步的阐释,但很可能涉及染色质的调控而引发了基因沉默。
表1 结肠癌细胞中发生突变及高甲基化的基因列表

遗传改变和表观遗传改变哪个先发生?
最近研究表明,尽管表观遗传调控的基因沉默在肿瘤发展过程中的任何阶段均可发生,但是在肿瘤形成的早期发生最频繁,如癌前病变阶段。研究发现家族性遗传的Wilms肿瘤病人,因基因印记的改变引起表观遗传异常有可能引起肾细胞异常的早期扩增,这种情况同样出现于结肠癌和其他癌症的早期。基因沉默与早期肺癌和乳腺癌中p16表达下调及前列腺癌中谷光苷肽-S-转移酶-π1(GSTP1)的表达下调相关,表观遗传基因沉默也与早期结肠癌一些基因的表达缺失相关。早期的表观遗传改变很有可能进一步促进细胞发生遗传异常,进一步加速了肿瘤的发展。例如,CDKN2A的沉默使得乳腺上皮细胞免于衰老,导致遗传的不稳定性以及肿瘤细胞的其他特性。而编码O6-甲基鸟嘌呤-DNA甲基转移酶的基因MGMT发生沉默,则使得细胞失去修复DNA鸟嘌呤核苷加合物的能力,从而出现特异性的遗传突变。
肿瘤细胞基因组研究表明表观遗传异常对于肿瘤的形成和发展有着重要的作用。这些研究表明,表观遗传的改变以及同遗传改变的相互作用使得肿瘤细胞依赖于各种致癌的信号途径,那么,仅仅通过表观遗传机制能否引起上述信号通路的异常?为了便于细胞克隆性扩增,转移前或者早期的肿瘤细胞是否因依赖于表观遗传调控引起某单一途径的失调,从而使肿瘤细胞避免正常的分化而进入异常增殖的状态?这种依赖性很有可能使得细胞内该信号途径获得后续突变,这又增加了细胞对异常信号途径的依赖性,最终导致了肿瘤的发展。另一种情况是,肿瘤细胞可能需要遗传和表观遗传同时发生改变而依赖信号途径异常。
Wnt途径异常的激活导致干细胞和祖细胞的扩增。在结肠癌中通常会发现肿瘤抑制基因APC发生突变而激活Wnt信号通路。目前认为在绝大多数的结直肠癌中,这些突变独自起作用就足以促使肿瘤的形成和发展。在培养的结肠肿瘤细胞中,通过恢复APC的功能而阻断Wnt信号途径的激活,便会导致细胞凋亡。肿瘤细胞依赖于单一的致癌基因调控的信号通路及对肿瘤抑制基因的失活表现出高敏感性,Weinstein认为上述的Wnt-APC信号途径就是一个重要的例证。
早期转移前的结肠肿瘤被称为异常隐窝病灶(ACF),有可能恶化成结肠癌。对这个阶段的肿瘤研究指出,在Wnt信号途径发生突变以前,表观遗传调控的机制有可能诱导了Wnt信号途径的激活(图1)。大多数ACF细胞中Wnt信号通路异常激活,但信号通路中的基因不发生突变,但是,这些细胞要发生恶化则需发生遗传改变。研究ACF细胞发现SFRP基因家族成员的启动子区域异常的甲基化。SFRP蛋白同frizzled蛋白有同源性,frizzled蛋白是Wnt信号蛋白的受体。SFRP在细胞膜上拮抗Wnt信号途径的激活。SFRP基因的启动子区域的甲基化不仅在ACFs细胞中检测到,而且在几乎所有的原位结肠癌细胞中都能检测到。并且,在SFRP基因沉默的结肠癌细胞中重新表达SFRP基因会阻断Wnt信号途径而引起细胞凋亡。重要的是,结肠癌细胞中Wnt信号通路下游的基因发生的失活突变和激活突变,被认为是结肠癌形成的原因。Wnt信号通路中的基因被称作“门控基因”(gatekeeper)。这些基因的失活被认为是结肠癌发生所必须的。
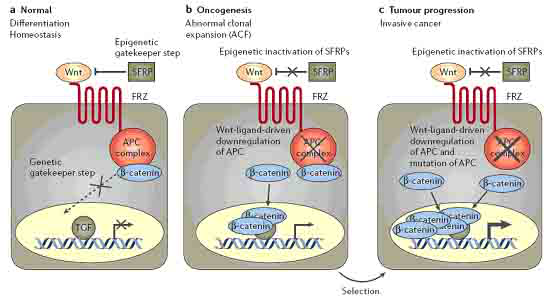
图1 通过基因沉默事件导致细胞依赖于Wnt信号通路。a|在正常的结肠上皮细胞中,分泌型的frizzled相关蛋白(SFRPs)的功能是与Wnt竞争性地同Wnt受体Frizzled结合,从而拮抗Wnt信号。当Wnt信号失活,腺瘤息肉病基因(APC)复合物磷酸化β-catenin,导致β-catenin降解。这便阻止了β-catenin的核内沉积,则不能激活转录因子(TCF),最终导致细胞进入分化并保持结肠上皮细胞处于动态平衡状态。b|通过表观遗传调控的基因沉默使得SFRP表达缺失,即“表观遗传调控门控基因”缺失,Wnt信号通路激活,促进细胞增殖及存活而不进入分化。c|持续性激活Wnt信号使得信号通路中的其他分子有可能发生突变,例如永久性失活APC复合物(图中为粗体×所示),即“遗传调控门控基因”缺失,进一步激活Wnt信号通路,从而促进肿瘤的发展。
因此,在结肠癌的早期,由于表观遗传调控的基因沉默,SFRP蛋白表达缺失,导致Wnt信号持续的激活。而由于Wnt信号激活促进结肠上皮干/祖细胞的异常扩增而抑制分化。随后这些细胞便高度依赖于过度激活的Wnt信号途径,于是便进一步要求Wnt信号途径下游的其他因子发生遗传突变,比如APC。而这些因子又可能进一步上调Wnt信号并促进肿瘤的发展。
肿瘤细胞的克隆性扩增和肿瘤的恶化是否要求表观遗传调控的“门控基因”(比如HCT116细胞中SFRP基因表达的激活)的丢失?Wnt信号下游的信号因子(比如β-catenin)的突变是否足以调控肿瘤发生发展这一整个过程?有两个证据说明肿瘤细胞克隆性扩增要求表观遗传的改变。一个是,在HCT116细胞中,将编码DNA甲基转移酶(DNMT)的基因缺失掉,会导致SFRP基因启动子去甲基化,SFRP基因重新表达,导致Wnt信号下调并诱导凋亡,尽管事实上这些细胞中表达激活形式的β-catenin,可能的原因是Wnt信号的丢失降低了细胞内β-catenin的表达量,包括突变的β-catenin(图1)。第二个证据是,在HCT116细胞和APC突变的细胞株中,外源性地重新表达SFRP基因,同样地,也能阻断Wnt信号通路并诱导凋亡。上述的证据强调了在驱动肿瘤发生的过程中表观遗传改变的重要性,逆转表观遗传的改变是潜在的干预和治疗肿瘤的手段。
在对小鼠和人类肿瘤细胞的研究基础上发现,另外一个受表观遗传调控沉默的基因HIC1与细胞依赖某一信号通路及肿瘤发展的早期阶段相关。在对染色体17p13.3的随机筛选中发现,高甲基化的HIC1在许多种人类癌症中缺失。在染色体上,HIC1基因位于肿瘤抑制基因p53的远端,它的缺失与p53的缺失和突变无关。HIC1是p53的靶基因,在早期乳腺癌和结肠癌中HIC1高甲基化。HIC1与干扰p53肿瘤抑制活动的一个复杂信号网络相关。HIC1途径的失活可能是肿瘤细胞依赖于某一致癌基因调控的信号通路及对肿瘤抑制基因的失活表现出高敏感这个理论的另一个例证(图2)。
HIC1通过抑制细胞应激敏感蛋白SIRT1基因的转录行使肿瘤抑制基因的功能,SIRT1是sirtuins组蛋白去乙酰化酶家族Ⅲ的一个成员。SIRT1对p53的转录后修饰(去乙酰化)抑制p53转录活性。HIC1和SIRT1还能形成一个复合物结合在SIRT1的启动子上。因此,在表观遗传调控引起HIC1失活的情况下, SIRT1的水平升高会促使p53的去乙酰化,这会削弱p53的功能,减少DNA的损伤引起的凋亡,最终导致肿瘤产生。
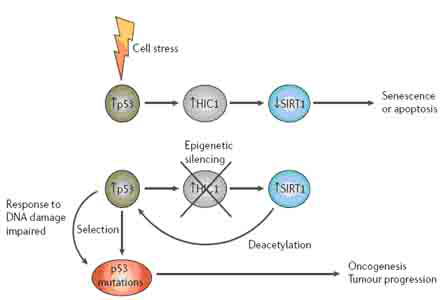
图2 表观遗传调控HIC1基因的沉默和DNA损伤应答。当正常细胞面临压力时,例如DNA损伤,p53便被激活,导致HIC1(癌症高甲基化基因1)转录表达。HIC1抑制编码应激敏感蛋白SIRT1的基因转录,这就使p53保持在乙酰化的活性状态。然而,在癌症发生的早期,表观遗传调控HIC1失活导致SIRT1的高表达,SIRT1使p53去乙酰化并削弱其功能,最终削弱DNA损伤引起的凋亡。这就使得在DNA损伤及突变沉积的情况下,细胞也能够增殖,破坏了正常的细胞周期调控并促进了肿瘤的发生发展。
对于SFRP和HIC1基因沉默的研究表明,表观遗传的改变在肿瘤发生过程中的作用包括两个方面。其一,表观遗传改变引起的基因沉默发生于肿瘤形成的早期(可能是干/祖细胞异常扩增阶段)。基因沉默使干细胞易于形成异常的克隆性扩增(图3)。在这一模型中,慢性炎症(伴随活性氧等有害物质产生)等促进肿瘤形成的致病因素引起强烈的细胞更新,用以修复组织。在更新过程中,表观遗传事件的发生导致某些基因转录抑制(可遗传),由此异常地激活或抑制细胞信号通路,引起干/祖细胞扩增。干/祖细胞异常扩增以正常细胞失去分化和成熟能力为代价的。其二,表观遗传改变引起的基因沉默与肿瘤的恶化相关。肿瘤恶化不仅依赖于遗传突变,还依赖于表观遗传改变的积累。这些表观遗传改变不仅在上皮细胞中持续发生,而且基质细胞也同时发生表观遗传改变。在肿瘤形成中,基质细胞的作用显得越来越重要。
在肿瘤发生过程中表观遗传异常作用与遗传异常作用同样重要,表观遗传异常使得肿瘤细胞依赖于异常的信号途径,因此它对于将来设计干预和治疗肿瘤的方法至关重要。很显然在体内遗传改变的逆转是很难实现的,但是表观遗传改变却能够被逆转。引起染色质变化的表观遗传改变,如启动子甲基化和组蛋白修饰引起转录水平异常,很可能是潜在的药物靶点,因为表观遗传沉默的基因可以被诱导重新表达。目前,能诱导沉默基因重新表达的药物已在测试中,比如DNA去甲基化药物和组蛋白去乙酰化酶抑制剂。被美国FDA批准的去甲基化药物5-aza-cytidine,对于白血病综合症前期脊髓发育不良的患者的恶性转化具有惊人的治疗和干预效果。
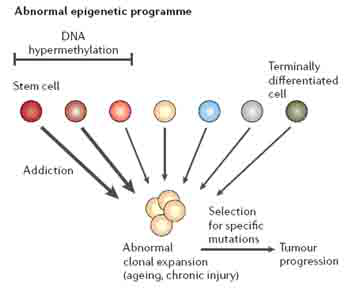
图3 表观遗传调控的基因沉默和肿瘤形成的关系。在正常成人上皮细胞更新过程中,在干/祖细胞常常发生基因沉默。当干/祖细胞分化成熟时,转录随之激活,这一稳态保证干/祖细胞在特定的上皮细胞系统中能沿着正常的分化路径分化(图中,从左至右)。在细胞慢性损伤或炎症时,适应性细胞更新压力迫使干/祖细胞更新用于修复,这一压力使得干/祖细胞原来沉默的基因启动子进一步发生DNA甲基化(图中,位于“DNA hypermethylation”bar下方的细胞)。这种永久性的沉默意味着当细胞克隆扩增时(粗箭头所示),转录不易被重新激活,干/祖细胞便进行异常增殖而不进入分化。类似的表观遗传过程也发生于支持异常上皮细胞扩增的基质细胞。整个过程导致良性肿瘤的形成,有恶化的风险。遗传或进一步表观遗传改变促进肿瘤恶化。表观遗传改变使干/祖细胞依赖于某一细胞信号通路进行异常扩增,信号通路其他分子进一步遗传突变或表观遗传改变便于肿瘤细胞克隆筛选。
机制
表观遗传改变如DNA甲基化是如何引起正常细胞向癌前病变细胞和肿瘤细胞转变呢?了解沉默过程中染色质的组成成分及其成因,对于肿瘤生物学研究及逆转基因沉默用于干预和治疗肿瘤至关重要。对于引发异常的启动子甲基化而导致肿瘤抑制基因的沉默已经有了一些线索。这些线索最初来自于对染色质生物学广阔领域的研究,尤其是对染色质生物学同肿瘤中基因沉默的交叉概念的研究。在胚胎发育和成熟细胞更新的过程中发生一系列的分子事件,比如干/祖细胞成熟的过程中标志性的基因处于活性形式而非转录抑制。这些标志性的事件的分子基础包括染色质重塑、基因核内定位以及组蛋白修饰。染色质重塑复合物中包括Trithorax和Polycomb蛋白家族,这两个家族的蛋白分别长效激活和抑制一些特定区域的基因表达,而这些基因与干/祖细胞分化成各种组织类型细胞是密切相关。
Polycomb复合物的研究(图4)为肿瘤细胞中异常的基因沉默的分子机制提供了证据。在果蝇或其他一些生物中,Polycomb复合物可以使一些基因持续的沉默,这种沉默机制对于胚胎基因和不同谱系基因的抑制是必须的。 Polycomb复合物PRC1(包含BMI1)、PRC2/3和PRC4(包含组蛋白甲基转移酶EZH2及其伴侣蛋白),在干/祖细胞及有干细胞特性的肿瘤细胞中处于活性形式。两个重要的肿瘤抑制基因,CDKN2A和CDNK2D在肿瘤细胞中频繁地高甲基化而沉默,它们是已知的BMI1间接或直接的靶基因。然而,复合物与基因间的相互作用同启动子甲基化引起的转录水平的基因沉默之间精确的联系还没有被研究清楚。
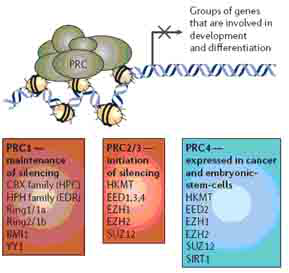
图4 polycomb抑制复合物(PRCs)的概况。PRCs有助于将核小体(图中黄色椭圆形所示)和附近的DNA(图中黑色链所示)组装到一起使得染色质转录受到抑制,介导下游基因的转录长期受到抑制。目前已确定的PRC有三种,这些复合物的蛋白成员在肿瘤细胞中常常过表达。PRC1在人类细胞中的功能是维持基因沉默,其成员见图中红色方框。PRC2/3起初始化基因沉默的作用,其成员见图中橙色方框。PRC4存在于胚胎干/祖细胞和癌症细胞中,其成员见图中蓝色方框。正常情况下,如果基因没有发生甲基化,而是由PRCs引起沉默会促进正常干细胞的功能,当干细胞成熟后这些基因的转录又被重新激活;但是在肿瘤细胞中,由于甲基化的增加促使这些基因持续的沉默,最终促进细胞的增殖。
在肿瘤发展的过程中,导致肿瘤细胞及它们的子代细胞中永久的基因沉默同启动子区域染色质组装的机制密切相关,尤其与关键的组蛋白氨基酸残基甲基化相关。肿瘤细胞中异常沉默基因和转录激活基因的组蛋白修饰是不同的。组蛋白H3的第9位赖氨酸和第27位赖氨酸的甲基化引起转录抑制。对链孢霉和拟南芥及哺乳动物X染色体的研究表明组蛋白甲基转移酶能招募DNMTs到基因的启动子区域。因此,在肿瘤中基因沉默的早期有可能发生组蛋白甲基化,随后引起启动子区域发生DNA甲基化。一些在结肠癌细胞中的实验性证据证实这个模型。
F.Rauscher及其同事提出一个很有吸引力的关于肿瘤形成过程中基因沉默序列事件模型。该研究中通过将特异性的转录抑制复合物与报告基因结合,在人细胞中诱导基因的瞬时沉默。这种结合能够定位出组蛋白转录抑制标志所在的区域,比如启动子区域组蛋白H3第9位赖氨酸的甲基化。当去除转录抑制剂,分离单个细胞来源的克隆,大多数克隆恢复激活靶基因的转录标志而丢失了转录抑制标志。然而,有些克隆中保持了启动子抑制标志,不能重新激活报告基因的转录,这些细胞通过启动子DNA的甲基化稳定地抑制转录。
相对于抑癌基因甲基化的成因,我们对其甲基化状态的维持了解得更多。多方面的研究表明,被沉默基因的启动子含有转录沉默的标志,除了CpG岛的甲基化外还包括组蛋白H3氨基酸的去乙酰化和甲基化。如组蛋白上第9和14位赖氨酸的去乙酰化、第9位赖氨酸的甲基化及第4位赖氨酸的甲基化程度降低。在这些抑制染色质的事件中,DNA甲基化似乎是基因沉默的主要因素。药物诱导的DNA去甲基化能使基因重新转录、转录激活标志出现(比如组蛋白第9、14位赖氨酸的乙酰化和第4个赖氨酸的甲基化)和基因沉默标志丢失(比如第9位赖氨酸的甲基化)。这些现象与早期的研究相一致,早期研究表明,用组蛋白去乙酰化酶的抑制剂处理肿瘤细胞,不能转录激活启动子高甲基化的肿瘤抑制基因,加入DNA去甲基化试剂才改变这一现象。
展望
表观遗传的改变和遗传改变一样,对于肿瘤的发生和发展非常重要。当细胞处于压力状态下,比如慢性损伤和炎症,发生一系列复杂的染色质修饰事件,可能通过表观遗传调控的机制,使一些重要的基因或整个基因网络锁定在异常的可遗传的转录抑制状态。这些机制使细胞依赖于重要的细胞调控途径引起不恰当的激活或紊乱。这种沉默被认为是“表观遗传调控的门控”阶段,基因沉默产生的环境有利于致癌基因或肿瘤抑制基因的选择性突变,从而促进肿瘤的发展。理解表观遗传调控的基因沉默的分子机制有利于促进通过逆转基因沉默过程的手段来干预和治疗肿瘤。


